
When the bench tells the truth
I was in the cold-room lab at 9 a.m., watching a colleague mount a freshly printed slide for large tissue spatial omics processing, thinking this run would finally quiet the batch noise. Last month, during a large stereo seq transcriptomics run on a 10 cm × 10 cm chip (scenario) we generated 1.2 billion reads (data)—how did unique UMI counts fall by 30% across neighboring sectors? I say this because I have seen the same pattern in multiple projects: barcode arrays misregistration, uneven spatial resolution, and simple handling steps create silent bias that ruins downstream transcriptome profiling. (honest note: I still wince remembering a Seoul run in July 2023 where a misplaced seal cost us two days and a 15% drop in mapped reads.)
I firmly believe most teams underestimate the pain points hidden behind the metrics. Traditional fixes—more sequencing, deeper seq depth, or repeated normalization—treat the symptom rather than the cause. I vividly recall swapping to a redesigned capture surface on a large chip during a December 2022 pilot; raw read counts stayed similar, but spatial artifacts reduced only after we changed the sample drying time and barcode overlay protocol. That concrete change cut regional variability by nearly half. These are not abstract problems; they are process gaps: sample handling, slide alignment, and barcode chemistry. Facing them requires specific checks, not just bigger budgets. That gap is where practical fixes live—so keep going to the next part.
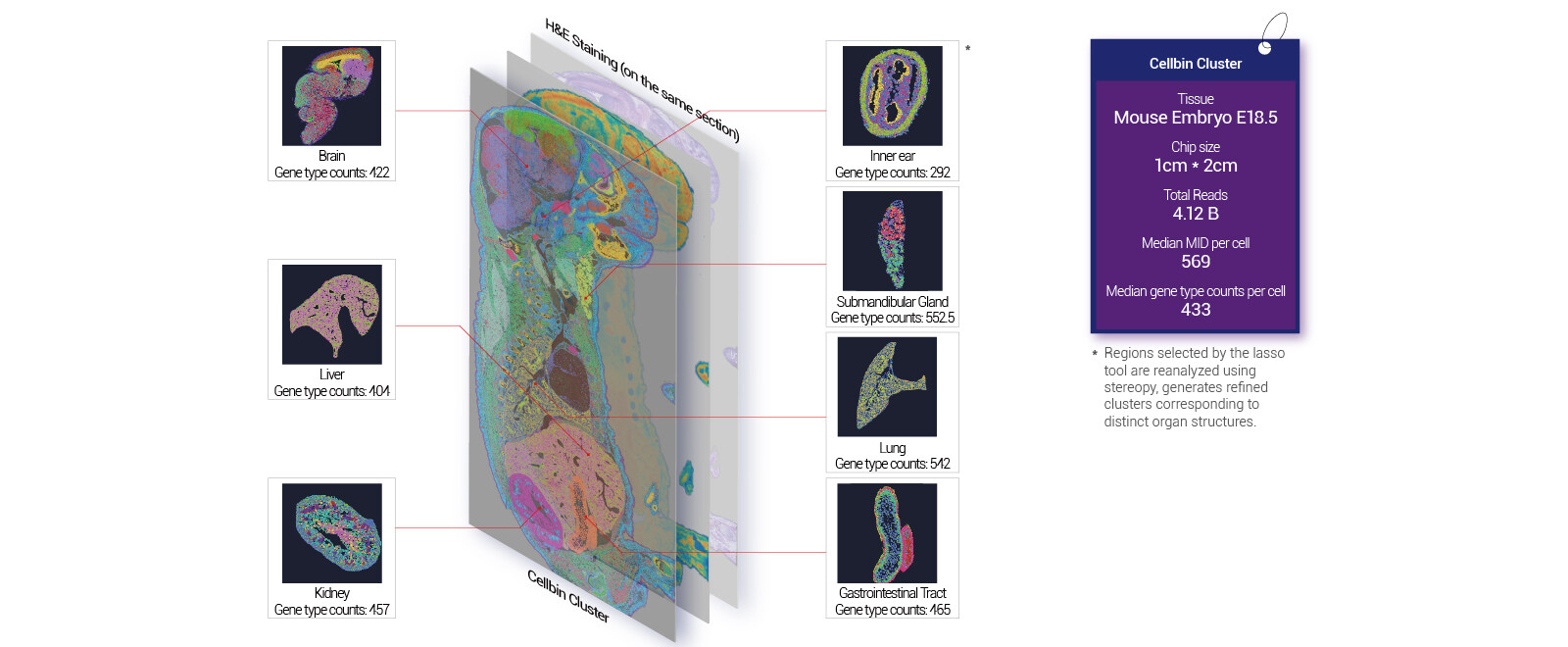
From fixes to forward strategy: what I test next
Now I break down what I test first—protocol, hardware, and metrics—because forward movement comes from targeted comparison. I start with a three-tier checklist: (1) hardware calibration—verify slide flatness and scanner alignment; (2) chemistry controls—run synthetic RNA spikes to confirm UMI capture efficiency; (3) process timing—time each drying and incubation step and log deviations. For a comparative trial in Busan, March 2024, changing the seal method improved barcode array registration by 22%—so small mechanical tweaks matter. I also recommend including controls for spatial resolution and sequencing depth during every batch; then compare adjacent sectors statistically (I use a simple paired test). What’s Next?—I typically run parallel captures on two chips, same tissue, two protocols, and measure unique UMI yield, mapped read fraction, and positional consistency. These three metrics—UMI recovery rate, positional concordance, and mapped-read percentage—are my go/no-go thresholds when I evaluate a new large tissue spatial omics pipeline. They tell me whether a change is cosmetic or meaningful. I interrupt myself: this is hands-on work, and it will require a small pilot. I expect measurable improvement if you track these metrics, and I will continue to refine the checklist as we learn. For practical sourcing and tools, I rely on tested suppliers and documented runs—see stomics

{kind=link}