
Introduction — a short, strange morning in the lab
I once walked into a morning that felt like a fairy tale gone slightly wrong: tiny glass vials glowing like lanterns, a stack of test reports and one uneasy client on the phone. In that hush I had to explain why a routine biological evaluation had turned into a three-month scramble. The numbers were stark — 22% repeat testing, two weeks extra on regulatory timelines — and the central question hovered: how do we find risks that hide beneath neat labels? (I still remember the smell of the solvent and the hum of the incubator.) I write from over 15 years of hands-on work in medical device toxicology consulting, and I want to share the kinds of patterns I watch for. Let’s move into the weeds with clear eyes and steady hands.

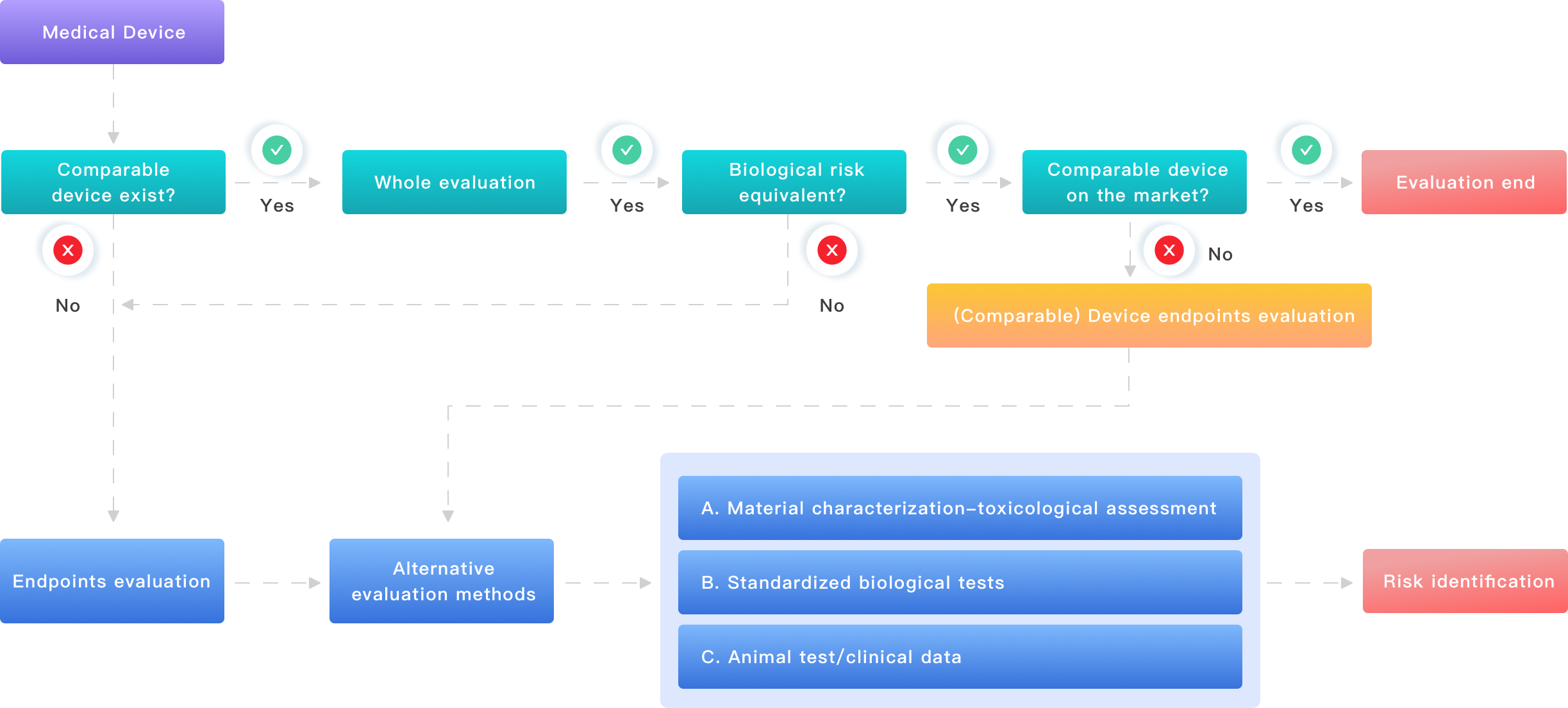
Traditional solution flaws: where the usual guidance breaks down
When teams ask me about toxicological risk assessment of medical devices, they expect a checklist. I learned early that checklists, by themselves, hurt more than help. Too often, manufacturers rely on generic pass/fail templates and ignore device context. That leads to missed extractables and leachables, or worse — misinterpreted cytotoxicity results. In April 2018, in my Boston lab, a client’s silicone Foley catheter flagged for borderline cytotoxicity. We traced it to a plasticizer not declared by a secondary supplier. The redesign cost the client roughly $28,400 in materials and retesting alone. That kind of avoidable expense stings and slows approvals.
Here’s the technical gap. Standards like ISO 10993 are essential, but they are not a substitute for chemical characterization tailored to your device. Labs often run standard assays without correlating them to actual clinical contact time or dynamic use. Systemic toxicity estimates get projected from short in vitro exposures when the device will sit in tissue for months. I press teams to map exposure scenarios — contact duration, surface area, release kinetics. Trust me, designing a risk matrix without those numbers is wishful thinking. The typical failure modes I see: incomplete material disclosure, inadequate extraction protocols, and low-sensitivity analytics that miss low-level leachables. Each hides costs and delays.
Why does this keep happening?
Because people assume test names equal safety. They do not. You must pair toxicology expertise with chemical and materials science. I often make time to review raw chromatograms (yes, the ones others skim) and the batch records from suppliers. That small extra step has stopped recalls and saved clients tens of thousands of dollars — and sometimes, months of wasted regulatory back-and-forth.
Future outlook: practical principles and what to change next
Looking forward, I advise teams to treat biological evaluation as a design-phase activity, not a final checkbox. In the next five years, the smartest programs will combine advanced chemical characterization with exposure modeling. That means using targeted GC–MS and LC–MS for extractables and leachables, pairing results with realistic simulants, and then translating those finds into dose estimates for systemic toxicity. I’ve begun recommending this approach to clients making polymeric implant coatings and ventricular assist device tubing in 2022 — we cut one project’s regulatory review time by five weeks simply by aligning test conditions to likely clinical use.
What’s Next — a few practical moves. First, insist on supplier transparency: get material data sheets and batch IDs. Second, run accelerated extraction plus real-use simulants. Third, adopt tiered analytics: broad screening, targeted confirmation, then quantitation. These steps help you avoid late-stage surprises — and yes, they require modest upfront spend. — pause for a second.
Three metrics I use when choosing a testing path
1) Exposure fidelity: Does the test mimic real contact time and body fluid? 2) Analytical sensitivity: Can the method quantify relevant leachables below thresholds of toxicological concern? 3) Traceability: Are supplier materials and batch numbers documented so a failure can be traced and fixed? I recommend these metrics because they measure what matters: patient safety, regulator confidence, and program cost. I see teams that ignore one of these metrics; they almost always pay later.
I speak from specific moments: a July 2016 weekend when I flagged a coated stent polymer that shed particles in saline at 37 °C; a November 2019 audit in Munich where missing supplier lot numbers led to a three-week delay. I recall the small wins as clearly as the pricey missteps. For teams in regulatory affairs and product development, these experiences matter — they change timelines, budgets, and reputation. If you want a practical partner to walk through this, consider established testing partners who combine toxicology, chemistry, and regulatory know-how. Wuxi AppTec Medical device testing
